





Abstract
The aim of the present study was to develop and validate a multitarget pyrose-quencing-based protocol for basic Chlamydia trachomatis genotyping directly from clinical samples and to characterize the distribution of genotypes among Slovenian sexually active population. The newly developed combination of assays that targets the variable domains VD-I and VD-IV of the C. trachomatisompA gene, was optimized and validated with 11 reference C. trachomatis strains and by comparison to complete ompA conventional sequencing. In addition, 183 clinical specimens which were previously diagnosed as C. trachomatis positive were evaluated by pyrosequencing. The pyrosequencing products showed a 100% match to corresponding sections of the respective conventional ompA sequences. Based on our results the most frequent genotype in urogenital samples was E (51.1%) followed by F (21.4%), G and K (6.9%), D (6.1%), H (3.8%), J (2.3%) and Ia and Ja (0.8%). In conjunctiva samples the genotype distribution was E (63.3%), D and F (13.3%), K (6.7%) and G (3.3%). Pyrosequencing thus proved itself to be a rapid method for C. trachomatis typing, which is important for better understanding the pathogenesis and epidemiology of this pathogen.
Introduction
Chlamydia trachomatis has been recognized as a major bacterial sexually transmitted infection and therefore it represents a significant global health issue (Gerbase et al., 1998). By immunotyping with polyclonal or monoclonal antibodies, C. trachomatis isolates can be divided into at least 15 prototypic serovars labeled A–K and L1–L3 based on the antigenic properties of the major outer membrane protein (MOMP) (Cabral et al., 2003). Traditional immunotyping methods that are applied for serotyping C. trachomatis require culturing of clinical samples and a large panel of monoclonal antibodies which make this approach difficult to use on routine bases (Zheng et al., 2007). However, C. trachomatis can be also typed according to nucleotide differences of the gene ompA, which encodes the MOMP, and serotypes can be similarly separated into genotypes (Millman et al., 2004) where in general types A–C cause trachoma, D–K cause ocular and genital diseases and L1–L3 cause lymphogranuloma venereum (Geisler et al., 2003). In epidemiological studies, C. trachomatis is mainly genotyped by ompA gene sequencing which requires long turnaround time, a lot of hands-on time and the cost is relatively high.
The DNA pyrosequencing method is a bioluminimetric de novo sequencing-technique which was first introduced in 1996 as a rapid and less expensive alternative to traditional Sanger DNA sequencing (Ronaghi et al., 1996). It has been developed for diverse applications, including single nucleotide polymorphism (SNP) detection, microorganism identification, and point mutations detection in antiviral or antimicrobial resistance genes as a strategy for molecular resistance testing (Luna et al., 2007; Marsh, 2007). However, best suited for SNP typing, with the recent development of optimized reagents, pyrosequencing can be also used for longer sequence analysis up to 70 nucleotides (Clarke, 2005). The main principle of pyrosequencing is to use a set of primers targeting conserved regions of a gene to obtain PCR products of all known genotypes and then to sequence the variable region between the selected primers (Ahmadian et al., 2006). This principle can be applied to the C. trachomatis ompA gene, since it has four variable domains designated as VD-I, VD-II, VD-III, and VD-IV (Yuan et al., 1989) that are flanked and interspaced by five constant domains (CD) (Stephens et al., 1988). VD-I and VD-II show the greatest amount of interserogroup sequence variation that codes for serovar specific epitopes. VD-IV on the other hand, is longer but more conserved and it codes for subspecies, serogroup, and highly conserved species specific antigenic determinants (Baehr et al., 1988; Stephens et al., 1988). The combination of VD-I, VD-II, and VD-IV thus seems to be suitable to achieve satisfactory resolution for genotype determination with the short sequences that are generated by the pyrosequencer.
The aim of the present study was to develop and validate a new multitarget pyrosequencing based protocol for basic C. trachomatis genotyping directly from clinical samples and to characterize the distribution of genotypes among symptomatic Slovenian sexually active population.
Materials and methods
Bacterial strains and clinical samples
The pyrosequencing assay was validated by using reference C. trachomatis strains D ;(IC-CAL 8), E ;(DK-20), F (MRC-301), G ;(IOL-238), H ;(UWA), I ;(UW12), J ;(UW36) K ;(UW31), L1 ;(440 L), L2 ;(434 B) and L3 ;(404L) which were obtained from the Institute of Ophthalmology (London, United Kingdom) and stored in liquid nitrogen. They were thawed and cultured on cycloheximide-treated McCoy cells (CRL 1696, ATCC) in shell vials as described before (Kese et al., 2005). The infected cells were harvested and used for DNA isolation immediately after cultivation. In addition, 183 clinical samples were enrolled in the study, including 74 urethral swabs from heterosexual men, 78 cervical swabs and 31 conjunctiva swabs placed in 1.5 ;mL of 2 ;M sucrose-phosphate (2SP) Chlamydia transport medium prepared in our laboratory. They were stored at −80 ;°C. The patient samples were consecutively collected during a 2-year period from 2007 to 2009 and had been previously found to be C. trachomatis positive by routine real-time PCR (COBAS® TaqMan® CT Test, v2.0; Roche, Germany) performed according to the manufacturer's instructions.
Moreover, additional bacterial isolates, which are listed in Table 1, were obtained from the culture collection of our Department of Bacteriology and included in this study to test the specificity of the PCR primers. Before testing, they were cultured on appropriate media and growth conditions with the following exceptions: Chlamydia pneumoniae, Chlamydia psittaci, and Mycoplasma genitalium were propagated in cell culture as appropriate. For total DNA isolation, five to ten bacterial colonies were suspended in 200 ;µL sterile buffer saline and 200 ;µL of infected cell cultures were prepared.
Bacterial species used to test the specificity of the VD-I and VD-IV ompA PCR primers
| Bacterial species | Source/strain |
| Chlamydia pneumoniae | Strain TW-183 |
| Chlamydia psittaci | ATCC VR-125 |
| Corynebacterium spp. | Clinical specimen |
| Enterococcus fecalis | Clinical specimen |
| Escherichia coli | ATCC 25922 |
| Klebsiella pneumoniae | Clinical specimen |
| Legionella pneumophila | Clinical specimen |
| Moraxella catarrhalis | Clinical specimen |
| Mycoplasma genitalium | Strain G37 |
| Mycoplasma hominis | Clinical specimen |
| Mycoplasma pneumoniae | Clinical specimen |
| Neiserria gonorrhoeae | Clinical specimen |
| Proteus mirabilis | Clinical specimen |
| Pseudomonas aeruginosa | ATCC 27853 |
| Staphylococcus aureus | ATCC 25923 |
| Streptococcus agalactiae | Clinical specimen |
| Streptococcus pyogenes | Clinical specimen |
| Ureaplasma spp. | Clinical specimen |
| Bacterial species | Source/strain |
| Chlamydia pneumoniae | Strain TW-183 |
| Chlamydia psittaci | ATCC VR-125 |
| Corynebacterium spp. | Clinical specimen |
| Enterococcus fecalis | Clinical specimen |
| Escherichia coli | ATCC 25922 |
| Klebsiella pneumoniae | Clinical specimen |
| Legionella pneumophila | Clinical specimen |
| Moraxella catarrhalis | Clinical specimen |
| Mycoplasma genitalium | Strain G37 |
| Mycoplasma hominis | Clinical specimen |
| Mycoplasma pneumoniae | Clinical specimen |
| Neiserria gonorrhoeae | Clinical specimen |
| Proteus mirabilis | Clinical specimen |
| Pseudomonas aeruginosa | ATCC 27853 |
| Staphylococcus aureus | ATCC 25923 |
| Streptococcus agalactiae | Clinical specimen |
| Streptococcus pyogenes | Clinical specimen |
| Ureaplasma spp. | Clinical specimen |
Bacterial species used to test the specificity of the VD-I and VD-IV ompA PCR primers
| Bacterial species | Source/strain |
| Chlamydia pneumoniae | Strain TW-183 |
| Chlamydia psittaci | ATCC VR-125 |
| Corynebacterium spp. | Clinical specimen |
| Enterococcus fecalis | Clinical specimen |
| Escherichia coli | ATCC 25922 |
| Klebsiella pneumoniae | Clinical specimen |
| Legionella pneumophila | Clinical specimen |
| Moraxella catarrhalis | Clinical specimen |
| Mycoplasma genitalium | Strain G37 |
| Mycoplasma hominis | Clinical specimen |
| Mycoplasma pneumoniae | Clinical specimen |
| Neiserria gonorrhoeae | Clinical specimen |
| Proteus mirabilis | Clinical specimen |
| Pseudomonas aeruginosa | ATCC 27853 |
| Staphylococcus aureus | ATCC 25923 |
| Streptococcus agalactiae | Clinical specimen |
| Streptococcus pyogenes | Clinical specimen |
| Ureaplasma spp. | Clinical specimen |
| Bacterial species | Source/strain |
| Chlamydia pneumoniae | Strain TW-183 |
| Chlamydia psittaci | ATCC VR-125 |
| Corynebacterium spp. | Clinical specimen |
| Enterococcus fecalis | Clinical specimen |
| Escherichia coli | ATCC 25922 |
| Klebsiella pneumoniae | Clinical specimen |
| Legionella pneumophila | Clinical specimen |
| Moraxella catarrhalis | Clinical specimen |
| Mycoplasma genitalium | Strain G37 |
| Mycoplasma hominis | Clinical specimen |
| Mycoplasma pneumoniae | Clinical specimen |
| Neiserria gonorrhoeae | Clinical specimen |
| Proteus mirabilis | Clinical specimen |
| Pseudomonas aeruginosa | ATCC 27853 |
| Staphylococcus aureus | ATCC 25923 |
| Streptococcus agalactiae | Clinical specimen |
| Streptococcus pyogenes | Clinical specimen |
| Ureaplasma spp. | Clinical specimen |
PCR and primers
The DNA from the C. trachomatis reference strains and from other bacterial isolates was extracted by using QIAamp DNA Mini Kit (Qiagen, Germany) according to the manufacturer's instructions. Genomic DNA from clinical samples was extracted from 100 ;µL of each sample using AMPLICOR CT/NG Specimen Preparation Kit (Roche) as described by the manufacturer.
The PCR was performed in multiple, separate reactions for the VD-I and VD-IV region of the C. trachomatis ompA gene using three amplification primer sets. Primer details and expected approximate amplicon lengths are shown in Table 2, while in Fig. ;1 the overall positioning of the primers is shown. Primer pairs omp-V1-F/omp-V1-Rb and omp-V1-Fb/omp-V1-R that amplify the whole VD-I region of C. trachomatis ompA were adopted from Jalal et al. (2007) (Table 2). Primers omp-V4-Fb and omp-V4-R that align to conserved nucleotides flanking the VD-IV region of the ompA gene, were newly designed using Assay Designer (Biotage, Sweden) and additionally checked for specificity with NCBI's blast.
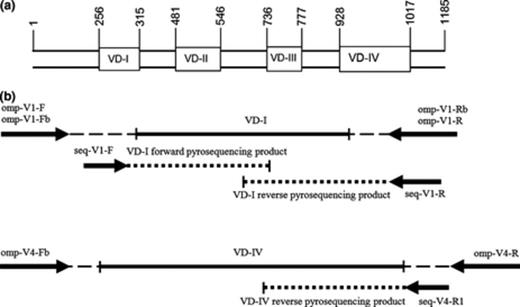
Overall procedure design: (a) schematic overview of the four VD regions of the Chlamydia trachomatisompA gene. Nucleotide positions are based on genotype F from Yuan et al. (1989); (b) positions of respective amplification and sequencing primers with their pyrosequencing products.
Amplification and pyrosequencing primers used in this study which target conserved regions flanking variable domains of the Chlamydia trachomatisompA gene
| Primer | Primer sequence | Source | Target | Approximate amplicon size (bp) |
| omp-V1-F | 5′-GACTTTGTTTTCGACCGTGTT-3′ | Jalal et al. (2007) | VD-I | 219 |
| omp-V1-Rb | 5′-biotin-ACA(AG)AATACATCAAA(AG)CGATCCCA-3′ | Jalal et al. (2007) | ||
| seq-V1-F | 5′-AAAC(AT)GATGTGAATAAAG-3′ | This study | ||
| omp-V1-Fb | 5′-biotin-GACTTTGTTTTCGACCGTGTT-3′ | Jalal et al. (2007) | VD-I | 219 |
| omp-V1-R | 5′-ACA(AG)AATACATCAAA(AG)CGATCCCA-3′ | Jalal et al. (2007) | ||
| seq-V1-R | 5′-ATCAAA(AG)CGATCCCA-3′ | This study | ||
| omp-V4-Fb | 5′-biotin-C(CT)TACATTGGAGTTAAATGG-3′ | This study | VD-IV | 225 |
| omp-V4-R | 5′-AATACCGCAAGATTTTCTAG-3′ | This study | ||
| seq-V4-R1 | 5′-TGTT(CT)AA(CT)TGCAAGGA-3′ | This study |
| Primer | Primer sequence | Source | Target | Approximate amplicon size (bp) |
| omp-V1-F | 5′-GACTTTGTTTTCGACCGTGTT-3′ | Jalal et al. (2007) | VD-I | 219 |
| omp-V1-Rb | 5′-biotin-ACA(AG)AATACATCAAA(AG)CGATCCCA-3′ | Jalal et al. (2007) | ||
| seq-V1-F | 5′-AAAC(AT)GATGTGAATAAAG-3′ | This study | ||
| omp-V1-Fb | 5′-biotin-GACTTTGTTTTCGACCGTGTT-3′ | Jalal et al. (2007) | VD-I | 219 |
| omp-V1-R | 5′-ACA(AG)AATACATCAAA(AG)CGATCCCA-3′ | Jalal et al. (2007) | ||
| seq-V1-R | 5′-ATCAAA(AG)CGATCCCA-3′ | This study | ||
| omp-V4-Fb | 5′-biotin-C(CT)TACATTGGAGTTAAATGG-3′ | This study | VD-IV | 225 |
| omp-V4-R | 5′-AATACCGCAAGATTTTCTAG-3′ | This study | ||
| seq-V4-R1 | 5′-TGTT(CT)AA(CT)TGCAAGGA-3′ | This study |
F, forward primer; R, reverse primer; seq, sequencing primer.
Amplification and pyrosequencing primers used in this study which target conserved regions flanking variable domains of the Chlamydia trachomatisompA gene
| Primer | Primer sequence | Source | Target | Approximate amplicon size (bp) |
| omp-V1-F | 5′-GACTTTGTTTTCGACCGTGTT-3′ | Jalal et al. (2007) | VD-I | 219 |
| omp-V1-Rb | 5′-biotin-ACA(AG)AATACATCAAA(AG)CGATCCCA-3′ | Jalal et al. (2007) | ||
| seq-V1-F | 5′-AAAC(AT)GATGTGAATAAAG-3′ | This study | ||
| omp-V1-Fb | 5′-biotin-GACTTTGTTTTCGACCGTGTT-3′ | Jalal et al. (2007) | VD-I | 219 |
| omp-V1-R | 5′-ACA(AG)AATACATCAAA(AG)CGATCCCA-3′ | Jalal et al. (2007) | ||
| seq-V1-R | 5′-ATCAAA(AG)CGATCCCA-3′ | This study | ||
| omp-V4-Fb | 5′-biotin-C(CT)TACATTGGAGTTAAATGG-3′ | This study | VD-IV | 225 |
| omp-V4-R | 5′-AATACCGCAAGATTTTCTAG-3′ | This study | ||
| seq-V4-R1 | 5′-TGTT(CT)AA(CT)TGCAAGGA-3′ | This study |
| Primer | Primer sequence | Source | Target | Approximate amplicon size (bp) |
| omp-V1-F | 5′-GACTTTGTTTTCGACCGTGTT-3′ | Jalal et al. (2007) | VD-I | 219 |
| omp-V1-Rb | 5′-biotin-ACA(AG)AATACATCAAA(AG)CGATCCCA-3′ | Jalal et al. (2007) | ||
| seq-V1-F | 5′-AAAC(AT)GATGTGAATAAAG-3′ | This study | ||
| omp-V1-Fb | 5′-biotin-GACTTTGTTTTCGACCGTGTT-3′ | Jalal et al. (2007) | VD-I | 219 |
| omp-V1-R | 5′-ACA(AG)AATACATCAAA(AG)CGATCCCA-3′ | Jalal et al. (2007) | ||
| seq-V1-R | 5′-ATCAAA(AG)CGATCCCA-3′ | This study | ||
| omp-V4-Fb | 5′-biotin-C(CT)TACATTGGAGTTAAATGG-3′ | This study | VD-IV | 225 |
| omp-V4-R | 5′-AATACCGCAAGATTTTCTAG-3′ | This study | ||
| seq-V4-R1 | 5′-TGTT(CT)AA(CT)TGCAAGGA-3′ | This study |
F, forward primer; R, reverse primer; seq, sequencing primer.
The reaction mixture of each of the VD-I PCR was composed of 1× AmpliTaq GOLD Buffer II (Applied Biosystems), 1.5 ;mM MgCl2 (AB), 200 ;nM of each forward and reverse primer (TIB MOLBIOL, Germany), 200 ;µM of each dNTP (AB), 1.5 ;U AmpliTaq GOLD Polymerase (AB), and molecular biology grade water (Qiagen) up to 45 ;µL. Five microliters of DNA template was added for a total volume of 50 ;µL. VD-I PCR was performed in a T3000 cycler (Biometra, Germany) under the following cycling parameters: 5 ;min at 95 ;°C followed by 50 cycles of 95 ;°C for 15 ;s, 54 ;°C for 30 ;s, 72 ;°C for 15 ;s and a final elongation step at 72 ;°C for 5 ;min. Finally, DNA from the same samples was also amplified by using a newly designed primer pair omp-V4-Fb/omp-V4-R in the VD-IV PCR (Table 2). The reaction mixture and cycling conditions were the same as those in the VD-I PCR, except for the annealing temperature, which was set to 51 ;°C, and the master mix contained 2.75 ;mM MgCl2. The PCR products were analyzed by electrophoresis on 2% agarose gel and ethidium bromide staining (1 ;µg mL−1). The DNA bands were visualized by UV transillumination on a Gel Doc 2000 imager (Bio-Rad).
Pyrosequencing and conventional ‘Sanger’ sequencing
Twenty microliters of PCR products was prepared for pyrosequencing by using the protocol for the vacuum prep tool according to the manufacturer's instructions (Biotage). Purified amplicons of the VD-I PCR were pyrosequenced in both directions using the newly designed sequencing primers seq-V1-F and seq-V1-R (Table 2). The sequencing primers seq-V1-F and seq-V1-R were constructed to align on opposite strands so that the resulting forward and reverse sequences would overlap at the ends. Finally, the amplicons corresponding to the VD-IV region were sequenced with the seq-V4-R1 sequencing primer. Pyrosequencing was performed on a PyroMarkID instrument (Biotage) with PyroMarkTM Gold Q96 SQA Reagents (Qiagen). All sequencing primers were designed using AssayDesigner (Biotage) and synthesized by TIB Molbiol (Germany); they are shown in detail in Table 2 and Fig. ;1. Specific dispensation orders of nucleotides for each respective assay were also developed. Genotype designation was performed with the use of IdentiFire software (Biotage) loaded with a custom built library and additionally checked by NCBI blast search.
As a confirmation method, conventional ‘Sanger’ sequencing was performed. PCR amplification was carried out using primers CT-1 and CT-5 designed from the first and the fifth conserved domain of the ompA gene, which gave DNA products of 1141 ;bp (Takourt et al., 2001). For the sequencing of the ompA gene, the PCR products were purified using a Wizard SV Gel and PCR Clean-Up system (Promega). As a template in the sequencing reactions 0.5–5 ;µL of the purified PCR products were amplified with the same primers using a BigDye® Terminator v3.1 Cycle Sequencing Kit (AB) according to the manufacturer's instructions. Sequencing reaction products were cleaned with a BigDye XTerminator® Purification Kit (AB) and sequenced in a 3500 Genetic Analyzer (AB) programmed to perform the long sequences protocol for obtaining maximum sequence data. The ompA sequences were analyzed and aligned with respective pyro-sequences using the clcmain workbench (CLC bio, Denmark) software.
Results
ompA VD-I and VD-IV PCR
The ompA VD-I and VD-IV fractions of C. trachomatis DNA were successfully amplified from all reference C. trachomatis strains and from 90.5% (67) of male genital, 82.1% (64) of female genital and 96.8% (30) of conjunctiva samples. All PCR products were of expected size and of suitable quantity. No unspecific amplification was observed in the VD-IV assay for all tested clinical samples. Furthermore, no cross-reactions were detected with DNA of other bacterial species by using newly designed primers in the VD-IV PCR assay.
Pyrosequencing
VD-I forward, VD-I reverse and VD-IV-pyrosequencing products were obtained from all reference C. trachomatis strains and all clinical samples that were positive in the respective PCR. VD-I forward and VD-I reverse-pyrosequencing products were used to construct an approximately 152 bases long consensus sequence that covers the whole VD-I. The consensus VD-I sequences and VD-IV pyrosequencing products showed a 100% match with adequate parts of the corresponding complete ompA ‘Sanger’ sequences, respectively.
Genotypes
In the presented study, the most prevalent genotype in urogenital samples was E (51.1%) followed by F (21.4%), G and K (6.9%), D (6.1%), H (3.8%), J (2.3%), and Ia and Ja (0.8%). In patients' conjunctiva samples the genotype distribution was E (63.3%), D and F (13.3%), K (6.7%), and G (3.3%). No sample with C. trachomatis DNA of genotype I, L1, L2 or L3 was detected in this study. Detailed results are shown in Table 3.
Distribution of Chlamydia trachomatis genotypes according to the sample origin
| Genotype | Urethral swab no. (%) | Cervical swab no. (%) | Conjunctiva swab no. (%) |
| D | 5 (7.5) | 3 (4.7) | 4 (13.3) |
| E | 40 (59.7) | 27 (42.2) | 19 (63.3) |
| F | 11 (16.4) | 17 (26.6) | 4 (13.3) |
| G | 4 (6.0) | 5 (7.8) | 1 (3.3) |
| H | 2 (3.0) | 3 (4.7) | 0 (0.0) |
| Ia | 1 (1.5) | 0 (0.0) | 0 (0.0) |
| J | 1 (1.5) | 2 (3.1) | 0 (0.0) |
| Ja | 0 (0.0) | 1 (1.6) | 0 (0.0) |
| K | 3 (4.5) | 6 (9.4) | 2 (6.7) |
| Total | 67 (100.0) | 64 (100.0) | 30 (100.0) |
| Genotype | Urethral swab no. (%) | Cervical swab no. (%) | Conjunctiva swab no. (%) |
| D | 5 (7.5) | 3 (4.7) | 4 (13.3) |
| E | 40 (59.7) | 27 (42.2) | 19 (63.3) |
| F | 11 (16.4) | 17 (26.6) | 4 (13.3) |
| G | 4 (6.0) | 5 (7.8) | 1 (3.3) |
| H | 2 (3.0) | 3 (4.7) | 0 (0.0) |
| Ia | 1 (1.5) | 0 (0.0) | 0 (0.0) |
| J | 1 (1.5) | 2 (3.1) | 0 (0.0) |
| Ja | 0 (0.0) | 1 (1.6) | 0 (0.0) |
| K | 3 (4.5) | 6 (9.4) | 2 (6.7) |
| Total | 67 (100.0) | 64 (100.0) | 30 (100.0) |
Distribution of Chlamydia trachomatis genotypes according to the sample origin
| Genotype | Urethral swab no. (%) | Cervical swab no. (%) | Conjunctiva swab no. (%) |
| D | 5 (7.5) | 3 (4.7) | 4 (13.3) |
| E | 40 (59.7) | 27 (42.2) | 19 (63.3) |
| F | 11 (16.4) | 17 (26.6) | 4 (13.3) |
| G | 4 (6.0) | 5 (7.8) | 1 (3.3) |
| H | 2 (3.0) | 3 (4.7) | 0 (0.0) |
| Ia | 1 (1.5) | 0 (0.0) | 0 (0.0) |
| J | 1 (1.5) | 2 (3.1) | 0 (0.0) |
| Ja | 0 (0.0) | 1 (1.6) | 0 (0.0) |
| K | 3 (4.5) | 6 (9.4) | 2 (6.7) |
| Total | 67 (100.0) | 64 (100.0) | 30 (100.0) |
| Genotype | Urethral swab no. (%) | Cervical swab no. (%) | Conjunctiva swab no. (%) |
| D | 5 (7.5) | 3 (4.7) | 4 (13.3) |
| E | 40 (59.7) | 27 (42.2) | 19 (63.3) |
| F | 11 (16.4) | 17 (26.6) | 4 (13.3) |
| G | 4 (6.0) | 5 (7.8) | 1 (3.3) |
| H | 2 (3.0) | 3 (4.7) | 0 (0.0) |
| Ia | 1 (1.5) | 0 (0.0) | 0 (0.0) |
| J | 1 (1.5) | 2 (3.1) | 0 (0.0) |
| Ja | 0 (0.0) | 1 (1.6) | 0 (0.0) |
| K | 3 (4.5) | 6 (9.4) | 2 (6.7) |
| Total | 67 (100.0) | 64 (100.0) | 30 (100.0) |
Discussion
Genotyping of C. trachomatis plays an important role in global epidemiological studies, as well as in local cases when questions of infection transmission or recurrence arise (Ikryannikova et al., 2010). In our study we have shown that pyrosequencing of C. trachomatis ompA gene could be applicable as a tool for genotyping this pathogen in epidemiological purposes. It is a user-friendly technology, cost-effective when a larger number of samples are being typed and rapid, which makes its use feasible. With the use of specific dispensation orders to minimize useless nucleotide dispensations, application of bidirectional approaches and targeting more than one area of a gene, the overall sequence length can be expanded and pyrosequencing products of 70–80 ‘good quality’ nucleotides can be readily achieved. However, inability to produce more than maximally 100 ;bp sequence reads still remains the main concern about liquid based pyrosequencing as a tool for bacterial genotyping, mainly due to apyrase wear off (Ronaghi et al., 1998). It is our belief that if key areas of a gene are carefully selected for sequencing, it is possible to achieve a satisfactory amount of sequence data to differentiate between genotypes. The possibility of using only short DNA sequences for genotyping C. trachomatis has been explored by other authors, using different methodological approaches. They used only portions of the ompA gene to type C. trachomatis with Oligonuclotide arrays (Zheng et al., 2007), Microsphere Suspention arrays (Huang et al., 2008), High Resolution Melting analysis (Li et al., 2010), Reverse Line Blot analysis (Molano et al., 2004), Multiplex Broad-Spectrum PCR-DNA-Enzyme Immunoassay coupled with Reverse Hybridization assay (Quint et al., 2007), and real-time PCR with genotype specific TaqMan probes (Jalal et al., 2007). In the present study we demonstrated the differentiation of C. trachomatis genotypes D-K by pyrosequencing the whole VD-I and a part of VD-IV region of the ompA gene. But it should be noted that genotypes L2a and L2b still cannot be discriminated between themselves and from L2. The resolution of the typing scheme would be higher if a pyrosequencing assay for the VD-II region could have been developed. Unfortunately, we were not successful in achieving a good assay due to miss priming of all tested sequencing primers (data not shown).
The major limitation of the pyrosequencing test as a genotyping tool is getting short sequences, with a maximum of 100 bases. Thus, pyrosequencing has been shown to provide limited discrimination for the identification of C. trachomatis intra-serotype sequence variations, especially for the predominating genotype E that in most studies comprises over 40% of the C. trachomatis infections (Jurstrand et al., 2010). A more discriminative method such as multilocus sequence typing or analysis of variable number tandem repeats should be considered. In cases with multiple serovar infections, pyrosequencing cannot resolve the genotypes involved directly. However, from the resulting pyrographs such infection could be suspected. A cloning approach of PCR products, transformation and subsequent sequencing of several plasmids would be needed.
In our study population a similar, however, slightly different distribution of genotypes was observed in urogenital samples from men and women, but with no statistically significant differences (Pearson's chi-squared test, α ;= ;0.05) for any of the genotypes under the assumption that each genotype will be equally represented in both genders. Genotype E is dominant in both genders with 59.7% and 42.2% for men and women, respectively, followed by genotype F represented with 16.4% in men and 26.6% in women. All other detected genotypes were found to be much less frequent. Although the difference between the two genders is the greatest for genotype E and F, the difference does not reach statistical significance (Pearson's chi-square test: genotype E, P ;= ;0.1122 α ;= ;0.05; genotype F, P ;= ;0.2568, α ;= ;0.05) for either of the genotypes. Our results are in concordance with several studies all over the world that assessed unselected or selected populations (Rodriguez et al., 1993; Ikehata et al., 2000; Millman et al., 2004; Gao et al., 2007; Mossman et al., 2008; Jurstrand et al., 2010; Machado et al., 2011) as shown in Table 4. In the study by Bandea et al. (2001) on pregnant Thai women genotype F was found to be dominant with 25% and E was only the fifth most common genotype with 9.3%. The studies by Petrovay et al. (2009) on high-risk Hungarian women (sex workers) and Molano et al. (2004) on a general Colombian women cohort interestingly found genotype D as the most prevalent genotype with 34.4% and 22.2%, respectively. However, in these two studies genotype E closely follows with 21.9% and 12.3%, respectively. It would seem that this difference might not be explained by group selection, since other studies performed on female sex workers (Lee et al., 2006; Gao et al., 2007), pregnant women (Ikehata et al., 2000), and women suffering urethritis or cervicitis (Zheng et al., 2007) found genotype E to be dominant. Regardless of the difference in the most prevalent genotype found in a specific study group, all studies including our own seem to be in concordance that genotypes D, E, and F are the most common genotypes in urogenital chlamydial infections as found by Naher & Petzoldt (1991) as early as 20 ;years ago.
Distribution of Chlamydia trachomatis genotypes among different study groups as found by other authors in recent years
| Typing method | Study group | No. of samples | Genotype distribution (%) | Country | References |
| Sequencing | Male and female patients | 237 bef. nvCT | E (47.3), F (17.3), D (13.5), K (8.9), J (4.2), Ia (3.0), H & G (2.5), Ba & E + F (0.4) | Sweden | Jurstrand et al. (2010) |
| 100 aft. nvCT | E (69.0), F (8.0), D (7.0), G (6.0), K (4.0), J & Ba (2.0), H & Ia (1.0) | ||||
| Sequencing | STD patients | 678 | E (39), F (21), G (11), D & Da & K (9), J (7), H (2), B & Ia (1) | Sweden | Lysén et al. (2004) |
| PCR + RFLP | Male and female patients | 203 | E (51.7), F (17.3), G (8.4), D (5.9), H & J (3.9), Dv & K (2.9), B & C (1.0), I & Ia (0.5) | France | Rodriguez et al. (1993) |
| Sequencing | Partners planning family | 507 | E (29.6), F (19.3), Ia (14.4), D (14.0), J (6.9), K (5.1), Ja (4.7), G (4.1), Ba (1), H (0.6), Ba/D (0.2) | USA | Millman et al. (2004) |
| Sequencing | Male and female patients | 163 | E (39.3), F (16.6), D (15.9), I (8.6), J (7.4), G (4.9), K (3.1), H (2.4), B (1.8) | Brazil | Machado et al. (2011) |
| Sequencing | STD patients | 189 | E (26.0), F (24.0), J (19.0%), D (13.0), H & K (6.0), G (4.0), B & I (1.0) | China | Yang et al. (2010) |
| PCR + RFLP | Female sex workers | 226 | E (27.9), F (23.5), G (12.4), D (11.1), K (6.6), mix (4.4), H (2.7), Ba, Un (1.3) | China | Gao et al. (2007) |
| PCR + RFLP Sequencing | Pregnant women | 207 | E (24.3), D (19.3), G (17.9), F (11.0), H & I (6.9), K (4.1), J (2.3), mix (1.8) | Japan | Ikehata et al. (2000) |
| Sequencing | N/A | 203 | E (42.36), F (23.65), G (16.26), D (9.36), H (4.43), mix (1.48), J (0.99), I & Ia & K (0.49) | Australia | Mossman et al. (2008) |
| Typing method | Study group | No. of samples | Genotype distribution (%) | Country | References |
| Sequencing | Male and female patients | 237 bef. nvCT | E (47.3), F (17.3), D (13.5), K (8.9), J (4.2), Ia (3.0), H & G (2.5), Ba & E + F (0.4) | Sweden | Jurstrand et al. (2010) |
| 100 aft. nvCT | E (69.0), F (8.0), D (7.0), G (6.0), K (4.0), J & Ba (2.0), H & Ia (1.0) | ||||
| Sequencing | STD patients | 678 | E (39), F (21), G (11), D & Da & K (9), J (7), H (2), B & Ia (1) | Sweden | Lysén et al. (2004) |
| PCR + RFLP | Male and female patients | 203 | E (51.7), F (17.3), G (8.4), D (5.9), H & J (3.9), Dv & K (2.9), B & C (1.0), I & Ia (0.5) | France | Rodriguez et al. (1993) |
| Sequencing | Partners planning family | 507 | E (29.6), F (19.3), Ia (14.4), D (14.0), J (6.9), K (5.1), Ja (4.7), G (4.1), Ba (1), H (0.6), Ba/D (0.2) | USA | Millman et al. (2004) |
| Sequencing | Male and female patients | 163 | E (39.3), F (16.6), D (15.9), I (8.6), J (7.4), G (4.9), K (3.1), H (2.4), B (1.8) | Brazil | Machado et al. (2011) |
| Sequencing | STD patients | 189 | E (26.0), F (24.0), J (19.0%), D (13.0), H & K (6.0), G (4.0), B & I (1.0) | China | Yang et al. (2010) |
| PCR + RFLP | Female sex workers | 226 | E (27.9), F (23.5), G (12.4), D (11.1), K (6.6), mix (4.4), H (2.7), Ba, Un (1.3) | China | Gao et al. (2007) |
| PCR + RFLP Sequencing | Pregnant women | 207 | E (24.3), D (19.3), G (17.9), F (11.0), H & I (6.9), K (4.1), J (2.3), mix (1.8) | Japan | Ikehata et al. (2000) |
| Sequencing | N/A | 203 | E (42.36), F (23.65), G (16.26), D (9.36), H (4.43), mix (1.48), J (0.99), I & Ia & K (0.49) | Australia | Mossman et al. (2008) |
STD, sexually transmitted disease; RFLP, restriction fragment length polymorphism; bef. nvCT, before discovery of new variant of C. trachomatis; aft. nvCT, after discovery of new variant of C. trachomatis; mix, mixed infection; N/A, not available; Un, unidentified; Dv, genotype D variant.
Distribution of Chlamydia trachomatis genotypes among different study groups as found by other authors in recent years
| Typing method | Study group | No. of samples | Genotype distribution (%) | Country | References |
| Sequencing | Male and female patients | 237 bef. nvCT | E (47.3), F (17.3), D (13.5), K (8.9), J (4.2), Ia (3.0), H & G (2.5), Ba & E + F (0.4) | Sweden | Jurstrand et al. (2010) |
| 100 aft. nvCT | E (69.0), F (8.0), D (7.0), G (6.0), K (4.0), J & Ba (2.0), H & Ia (1.0) | ||||
| Sequencing | STD patients | 678 | E (39), F (21), G (11), D & Da & K (9), J (7), H (2), B & Ia (1) | Sweden | Lysén et al. (2004) |
| PCR + RFLP | Male and female patients | 203 | E (51.7), F (17.3), G (8.4), D (5.9), H & J (3.9), Dv & K (2.9), B & C (1.0), I & Ia (0.5) | France | Rodriguez et al. (1993) |
| Sequencing | Partners planning family | 507 | E (29.6), F (19.3), Ia (14.4), D (14.0), J (6.9), K (5.1), Ja (4.7), G (4.1), Ba (1), H (0.6), Ba/D (0.2) | USA | Millman et al. (2004) |
| Sequencing | Male and female patients | 163 | E (39.3), F (16.6), D (15.9), I (8.6), J (7.4), G (4.9), K (3.1), H (2.4), B (1.8) | Brazil | Machado et al. (2011) |
| Sequencing | STD patients | 189 | E (26.0), F (24.0), J (19.0%), D (13.0), H & K (6.0), G (4.0), B & I (1.0) | China | Yang et al. (2010) |
| PCR + RFLP | Female sex workers | 226 | E (27.9), F (23.5), G (12.4), D (11.1), K (6.6), mix (4.4), H (2.7), Ba, Un (1.3) | China | Gao et al. (2007) |
| PCR + RFLP Sequencing | Pregnant women | 207 | E (24.3), D (19.3), G (17.9), F (11.0), H & I (6.9), K (4.1), J (2.3), mix (1.8) | Japan | Ikehata et al. (2000) |
| Sequencing | N/A | 203 | E (42.36), F (23.65), G (16.26), D (9.36), H (4.43), mix (1.48), J (0.99), I & Ia & K (0.49) | Australia | Mossman et al. (2008) |
| Typing method | Study group | No. of samples | Genotype distribution (%) | Country | References |
| Sequencing | Male and female patients | 237 bef. nvCT | E (47.3), F (17.3), D (13.5), K (8.9), J (4.2), Ia (3.0), H & G (2.5), Ba & E + F (0.4) | Sweden | Jurstrand et al. (2010) |
| 100 aft. nvCT | E (69.0), F (8.0), D (7.0), G (6.0), K (4.0), J & Ba (2.0), H & Ia (1.0) | ||||
| Sequencing | STD patients | 678 | E (39), F (21), G (11), D & Da & K (9), J (7), H (2), B & Ia (1) | Sweden | Lysén et al. (2004) |
| PCR + RFLP | Male and female patients | 203 | E (51.7), F (17.3), G (8.4), D (5.9), H & J (3.9), Dv & K (2.9), B & C (1.0), I & Ia (0.5) | France | Rodriguez et al. (1993) |
| Sequencing | Partners planning family | 507 | E (29.6), F (19.3), Ia (14.4), D (14.0), J (6.9), K (5.1), Ja (4.7), G (4.1), Ba (1), H (0.6), Ba/D (0.2) | USA | Millman et al. (2004) |
| Sequencing | Male and female patients | 163 | E (39.3), F (16.6), D (15.9), I (8.6), J (7.4), G (4.9), K (3.1), H (2.4), B (1.8) | Brazil | Machado et al. (2011) |
| Sequencing | STD patients | 189 | E (26.0), F (24.0), J (19.0%), D (13.0), H & K (6.0), G (4.0), B & I (1.0) | China | Yang et al. (2010) |
| PCR + RFLP | Female sex workers | 226 | E (27.9), F (23.5), G (12.4), D (11.1), K (6.6), mix (4.4), H (2.7), Ba, Un (1.3) | China | Gao et al. (2007) |
| PCR + RFLP Sequencing | Pregnant women | 207 | E (24.3), D (19.3), G (17.9), F (11.0), H & I (6.9), K (4.1), J (2.3), mix (1.8) | Japan | Ikehata et al. (2000) |
| Sequencing | N/A | 203 | E (42.36), F (23.65), G (16.26), D (9.36), H (4.43), mix (1.48), J (0.99), I & Ia & K (0.49) | Australia | Mossman et al. (2008) |
STD, sexually transmitted disease; RFLP, restriction fragment length polymorphism; bef. nvCT, before discovery of new variant of C. trachomatis; aft. nvCT, after discovery of new variant of C. trachomatis; mix, mixed infection; N/A, not available; Un, unidentified; Dv, genotype D variant.
Among conjunctiva swab samples of adults and newborn babies we noticed genotype E to be even more dominant (63.3%); this could be the result of a smaller patient group. However, Gallo Vaulet et al. (2010) found a significantly higher frequency of genotype E in the cases of ophthalmia neonatorum. This finding could not only be due to the genital predominance of genotype E and transmission to the conjunctiva, but also other factors might be implicated in the pathogenesis of this genotype. Therefore, a larger study should be performed to shed more light on the distribution of C. trachomatis genotypes in ocular samples of newborn babies and adults.
The pyrosequencing method is rapid, it takes only 15 ;min for the preparation step and maximally 2 ;h for the sequencing process of 96 samples. Its high through-put makes it economically effective. This method might be a valuable tool for genotyping of C. trachomatis, and still more studies should be made.
Acknowledgement
The authors would like to express their gratitude to Luka Fajs, BSc for his technical assistance with conventional sequencing.
References

{kind=link}